Gaschromatographie (GC)
Definition
- Chromatographisches Verfahren, bei dem als
mobile Phase statt einer Flüssigkeit ein Gas eingesetzt wird.
Messprinzip
- Die GC ist in erster
Linie eine Verteilungschromatographie. Allerdings muss auch der Dampfdruck
der Substanzen berücksichtigt werden.
Prinzip der GC
- Die Probe kann als Gas, Flüssigkeit oder
gelöster Feststoff eingesetzt werden.
Für Flüssigkeiten und gelöste Feststoffe gilt allerdings die Einschränkung,
dass sie sich unter den gegebenen Bedingungen von ca. max. 400
°C verdampfen lassen.
- Die Probe wird, sofern sie nicht bereits
gasförmig vorliegt, im Injektor verdampft. Je nach Dampfdruck geht dies schneller oder
langsamer.
- Die verdampfte Probe wird
nun einem inerten Trägergas beigefügt und durch die Trennsäule gespült.
Hier erfolgt nun die Trennung nach Lipophilie bzw. Hydrophilie.
- Die Auswertung erfolgt anhand der die Säule
verlassenden Substanzen mittels eines
Detektors (äußeres Chromatogramm)
Trägergas
- Das Trägergas, die mobile Phase der GC,
dient dem Transport der Substanzen durch
den Gaschromatographen. Ein ideales Trägergas sollte folgende Eigenschaften
aufweisen:
- chemisch inert
- gute Trenneigenschaften (van-Deemter-Gleichung)
- preiswert
- nicht brennbar
| Trägergas |
Vorteile
|
Nachteile
|
| Wasserstoff
|
preiswert, gute Trenneigenschaften |
brennbar |
| Helium
|
gute Trenneigenschaften |
teuer |
| Stickstoff
|
preiswert |
schlechte Trenneigenschaften |
| Argon
|
gute Trenneigenschaften |
teuer |
| Kohlendioxid |
obsolet |
- Die Zufuhr des Trägergases wird mittels
Ventilen geregelt. Je nach der Einstellung
kann man die Strömungsgeschwindigkeit variieren.
- Je schneller das
Gas strömt, desto kürzer werden zwar die Analysenzeiten, jedoch verschlechtert
sich auch die Trennleistung, da weniger Zeit für die Einstellung
des Verteilungsgleichgewichtes bleibt.
- In der GC werden keine Gemische von Trägergasen eingesetzt.
Injektor
- Sind die Proben nicht gasförmig, so müssen
sie erst in den gasförmigen Zustand
überführt werden. Dies geschieht im Injektor, bzw. im vorderen Teil
der Säule.
- Split-Injektor
- Die Probe wird mittels einer
Mikroliterspritze durch ein Teflon-Septum in einer
Kammer injiziert, dort verdampft und dem Trägergasstrom zugefügt.
- Vor dem Verlassen des Injektors wird der
Gasstrom mit Hilfe eines Ventils geteilt.
Ein Teil geht auf die Trennsäule, der andere wird verworfen.
- Durch dieses Verfahren erhält man eine
zusätzliche Verdünnung der Probe, eine
Überladung der Säule wird also vermieden. Negativ ist jedoch, dass ein
Teil der Probe ungenutzt verloren geht, was sich vor allem bei nur kleinen
Probenmenge bemerkbar macht.
- On-Column-Injektor
- Die Probe wird durch ein Ventil direkt auf
die Säule gespritzt und verdampft dort
langsam. Der Injektor dient also allein zur Einbringung der der
Probe und Zumischung des Trägergases. Um eine Überladung der Säule zu vermeiden,
dürfen nur wesentlich kleinere Probenmengen, als beim Split-Injektor
aufgetragen werden, allerdings wird die Probe vollständig genutzt,
was besonders bei nur kleinen Probenmengen von Vorteil ist.
- Die Verdampfbarkeit der Probe stellt eine
Grenze der Anwendbarkeit der GC dar.
Ist eine Probe unter den gegebenen Bedingungen nicht flüchtig oder nicht
stabil, so kann sie nicht ohne weiteres untersucht werden. In einigen
Fällen kann die Probe jedoch durch Derivatisierung doch noch in eine
bestimmbare Form überführt werden.
- Durch Veresterung, Veretherung, Silylierung
und andere Verfahren, kann z.B. die
Flüchtigkeit erhöht, die Zersetzung vermieden, eine Trennung von Enantiomeren
ermöglicht oder die Detektierbarkeit (z.B. für ECD) erreicht werden.
- Beim Einsatz gasförmigere Proben kann die
Head-Space-Technik angewandt werden.
Eine flüssige Probe wird hierbei in einem geschlossenen Gefäß erwärmt.
Zwischen dem Luftraum über der Flüssigkeit und der flüssigen Phase stellt
sich ein Gleichgewicht ein. Die Probe wird nun aus dem Luftraum über
der Probe entnommen und der GC zugeführt.
Säule
- In der GC werden zwei Säulentypen verwandt, man unterscheidet:
- Gepackte Säulen
- Gepackte Säulen sind 0,5 - 8 m lang und haben Innendurchmesser von 2 - 4 mm.
- Als Packungsmaterial dienen Adsorbentien wie Kieselgel oder Aluminiumoxid. Kieselgur wird als Trägermaterial für schwer verdampfbare Flüssigkeiten (Trennflüssigkeit) benutzt.
- Vorteilhaft bei gepackten Säulen ist ihre Belastbarkeit mit höheren Substanzmengen, allerdings ist ihre Trennleistung deutlich geringer als die von Kapillarsäulen.
- Kapillarsäulen
- Kapillarsäulen sind 15 - 100 m lang und haben Innendurchmesser von ca. 0,1 mm.
- Es werden zwei Untertypen unterschieden: Dünnfilmsäulen und Dünnschichtsäulen.
- Bei den Dünnfilmsäulen
ist auf die Kapillarinnenseite eine Trennflüssigkeit aufgebracht. Da sie nicht zusätzlich fixiert ist, neigen Dünnfilmsäulen zum "Ausbluten", dem Austritt der Trennflüssigkeit aus der Säule während der Aufnahme des
Chromatogramms. Dies setzt ihre Haltbarkeit deutlich herab. Die Belastbarkeit von Dünnfilmsäulen liegt bei ca. 0,001 µl.
- Dünnschichtsäulen
enthalten auf der Kapillarinnenseite eine dünne Trägerschicht aus z.B. Aluminiumoxid oder Kieselgel. Auf diese Trägerschicht ist eine geringe Menge einer Trennflüssigkeit aufgetragen. Diese ist dort weitestgehend immobilisiert, so dass
Dünnschichtsäulen praktisch nicht "ausbluten" und deutlich länger halten als Dünnfilmsäulen. Die Belastbarkeit von Dünnschichtsäulen beträgt ca. 0,01 - 0,1 µl.
- Als Trennflüssigkeiten kommen zahlreiche Verbindungen zum Einsatz, die sich in ihrer Lipophilie bzw. Hydrophilie unterscheiden. Gemeinsam ist ihnen ihre hohe Molmasse und damit ihr niedriger Dampfdruck. Meist handelt
es sich um Polymere. Eingesetzte Stoffklassen sind Alkane / Paraffine, Silikonöle oder Polyethylenglykole.
- Säulen ohne (immobilisierte) Trennflüssigkeiten kommen heute praktisch nur noch bei der Analyse von Gasgemischen zum Einsatz.
Detektor
- Die drei am häufigsten verwendeten
Detektoren sind:
- Flammenionisationsdetektor (FID)
- Der von der Säule kommende Gasstrom
wird in einer Knallgasflamme verbrannt. Zwischen den Brenner und
einer Kollektorelektrode wird eine Spannung angelegt. Durch in der
Flamme bei Elution einer Verbindung entstehende Ionen und Radikale
wird ein Stromfluss erzeugt, der als Messsignal
registriert wird.
- Die Nachweisgrenze liegt bereits
relativ niedrig.
- Wärmeleitfähigkeitsdetektor (WLD)
- Eine mit einem Temperaturfühler
versehene Kammer wird allein vom Trägergas durchströmt.
Eine andere, ansonsten baugleiche, Kammer vom Gasstrom aus der
Säule. Gelangt aus der Säule ebenfalls nur das Trägergas in diese
Zelle, so registrieren beide Fühler die gleiche Wärmeableitung.
Befinden sich im Gasstrom der Säule jedoch Substanzen, so ändert
sich die Wärmeleitfähigkeit des Gases und es tritt eine
Temperaturdifferenz zwischen den Messfühlern auf. Diese wird als
Messsignal aufgenommen.
- Die Nachweisgrenze liegt etwas
höher, als beim FID.
- Eingesetzt wird der WLD vor allem bei der Verwendung von Wasserstoff
als Trägergas, seltener auch noch bei Helium. Bei anderen Trägergasen spielt er praktisch keine Rolle.
- Elektroneneinfangdetektor (EED)
- Synonym
- electron capture detector (ECD)
- Der Trägergastrom wird durch einen b-Strahler (z.B. 3H, 63Ni) ionisiert.
- Diese Ionisierung führt zu einem
Grundstrom, der zwischen zwei Elektroden gemessen
wird. Treten Substanzen aus der Säule, die Elektronen einfangen
können, so wird der Grundstrom abgeschwächt. Diese wird
messtechnisch aufgezeichnet und später ausgewertet.
- Die Nachweisgrenze liegt sehr niedrig.
- Neben diesen drei Detektoren ist ferner die
GC-MS von Bedeutung. Hier wird die
Gaschromatographie mit der Massenspektrometrie gekoppelt.
Chromatogramm
- Die genannten Detektoren liefern ein Signal,
das der Schreiber in einen Peak im
Chromatogramm umwandelt. Diese Signale werden gegen die Zeit aufgetragen.
-
Die Zeit nach der Injektion der Probe bis zum Auftreten des Signals
wird als Gesamtretentionszeit tm+s bezeichnet. Sie entspricht der
Aufenthaltszeit der Substanz in
stationärer und mobiler Phase.
- Substanzen, die
keinerlei Wechselwirkungen mit der stationären Phase zeigen, werden trotzdem
nicht zum Zeitpunkt t = 0 registriert, da auch sie erst den Weg vom Injektor
durch die Säule zum Detektor zurücklegen müssen. Diese Zeit, die der
Aufenthaltszeit der mobilen Phase im System entspricht, wird als Totzeit
bezeichnet. Die Totzeit einer BPI-Säule kann z.B. durch die Injektion
von Methan bestimmt werden.
Trennstufenzahl / HETP
- Zur Charakterisierung der Trennleistung
einer Säule wird die Trennstufenzahl
n, bzw. die Trennstufenhöhe (HETP, "height equivalent of a theoretival
plate") verwendet. Sie ist eine für
jede Substanz charakteristische Größe, die von der Säule und den Trennbedingungen
abhängt.
- Stellt man sich die Säule als Raum mit
vielen kleinen Kammern vor, so strömt das Gas mit der Substanz in jede
Kammer, wo sich ein Verteilungsgleichgewicht zwischen Gasraum und der
Trennflüssigkeit einstellt. Je mehr Kammern es gibt, desto besser ist die
Trennleistung und desto höher ist die Trennstufenanzahl bzw. desto
niedriger ist die HETP.
- Die Trennstufenzahl lässt sich aus der
Retentionszeit berechnen:
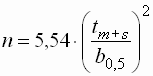
| n |
: |
Trennstufenzahl |
| tm+s |
: |
Gesamtretentionszeit |
| b0,5 |
: |
Halbwertsbreite |
- Für die Trennstufenhöhe ergibt sich
danach:
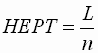
| HEPT |
: |
Trennstufenhöhe
("height equivalent of a theoretical plate") |
| L |
: |
Länge der Säule |
| n |
: |
Trennstufenzahl |
Van-Deemter-Gleichung
- Die van-Deemter-Gleichung wird als
Grundgleichung der GC bezeichnet. Sie stellt
den Zusammenhang zwischen Trennstufenhöhe und Strömungsgeschwindigkeit
dar.
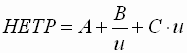
| HEPT |
: |
Trennstufenhöhe ("height
equivalent of a theoretical plate") |
| A |
: |
Streudiffusion |
| B |
: |
Longitudinaldiffusion |
| C |
: |
Massenübertragung |
| u |
: |
Strömungsgeschwindigkeit |
- Die Streudiffusion, auch als
Eddy-Diffusion bezeichnet, tritt durch unregelmäßig gepackte Säulen mit
unterschiedlichen Strömungsgeschwindigkeiten in den Hohlräumen auf. Sie
sorgt für eine Zonenverbreiterung. Je größer die Hohlräume zwischen den
einzelnen Partikeln der stationären Phase ist, desto stärker ist der durch
die Streudiffusion entstehende negative Effekt. Ideal ist folglich eine
homogene Packung der Säule mit möglichst kleinen Teilchen.
- Die Longitudinaldiffusion bezeichnet
die Diffusion in Richtung der Trennstrecke. Sie kann durch hohe
Trägergasgeschwindigkeiten klein gehalten werden.
- Die Massenübertragung bezieht sich
auf die Geschwindigkeit mit der sich der Gleichgewichtszustand einstellt.
- Bei der Kapillar-GC sind Streu- und
Longitudinaldiffusion zu vernachlässigen.
- Da die Terme B und C durch die
Strömungsgeschwindigkeit (u) gegensätzlich beeinflusst werden, muss das
Optimum der Trägergasgeschwindigkeit durch Vorversuche ermittelt werden.
Trägt man die erhaltenen Werte für HEPT gegen die
Strömungsgeschwindigkeit auf, so erhält man Kurven wie:
- BILD
- Jedes Trägergas weist einen unterschiedlichen
Kurvenverlauf auf. Anhand der Kurve werden die unterschiedlichen
Trenneigenschaften der Gase sichtbar. So zeigt Stickstoff
nur ein schmales Minimum, weshalb sich Stickstoff nur schwer in
der GC einsetzen lässt.
- Die van-Deemter-Gleichung ist weitgehend
auch auf die HPLC übertragbar.
Qualitative Bestimmung
- Durch Vergleich der Retentionszeit mit einer
Referenz ist eine Identifizierung von Substanzen
möglich. Die Retentionszeit allein kann jedoch
nicht aus Tabellen entnommen und verglichen werden, da sie von zahlreichen
Faktoren abhängig ist:
- Art und Länge der Säule
- Operator
- Art des Trägergases
- Temperatur von Injektor, Detektor und
Säule
- Art des Detektors
- Strömungsgeschwindigkeit
- Um diese Faktoren auszumitteln, wird die
relative Retentionszeit gebildet:
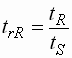
| trR |
: |
relative Retentionszeit |
| tR |
: |
Retentionszeit der Referenz |
| tS |
: |
Retentionszeit der Substanz |
- Des weiteren
wurde von Kovats eine Methode eingeführt, die diese Faktoren berücksichtigt.
Der sogenannte Kovats-Index erlaubt die Identifizierung einer
Substanz. Allerdings weisen oftmals verschiedene Substanzen den gleichen
oder einen sehr ähnlichen Kovats-Index auf.
- ....
- Bei einem beliebigen Trennsystem wird zuerst
eine Mischung verschieden langer
n-Alkane getrennt. Der Logarithmus ihrer Nettoretentionszeit wird gegen
die Kettenlänge mal Faktor 100 aufgetragen. Man erhält eine Gerade.
- Die Retentionszeit der Probe wird nun unter
gleichen Bedingungen ermittelt. Zu der
nun gemessenen Zeit wird anhand der Gerade eine theoretische
Kettenlänge interpoliert. Dieser kann nun anhand von Tabellen eine
Substanz zugeordnet werden.
- Sollten mehrere Substanzen in Frage kommen, so kann eine Identifizierung, z.B. durch die Zugabe einer geringen Menge der erwarteten Substanz und die Aufnahme eines weiteren Chromatogramms unter etwas veränderten
Bedingungen, durchgeführt werden. Sind im erhaltenen Chromatogramm keine neuen Peaks zu erkennen und ist der Peak des zu identifizierenden Stoffs nun größer, so kann von Identität ausgegangen werden.
- Der Kovats-Index bezieht sich auf eine
isotherme Arbeitsmethode, bei Temperaturprogrammen versagt er.
- Die Berechnung des Kovats-Index ist
ebenfalls möglich:
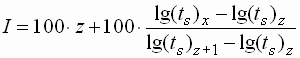
| I |
: |
Kovats-Index |
| x |
: |
Wert der gesuchten
Substanz |
| z |
: |
Werte des Alkans mit
kleinerer Retentionszeit |
| z+1 |
: |
Werte des Alkans mit
nächsthöherer Retentionszeit |
Quantitative Bestimmung
- Die quantitative Bestimmung erfolgt gegen
Standards. Zum einen wird ein externer
Standard benutzt, der den zu bestimmenden Stoff in bekannter Konzentration
enthält, zum anderen ein interner Standard, der einen beliebigen
Stoff in einer bestimmten Konzentration enthält. Der interne Standard
wird sowohl zur Proben- als auch zur Referenzlösung gegeben und ermöglicht
später die Beseitigung falscher Ergebnisse durch Dosierungenauigkeiten
bei der Substanzapplikation in den Automaten. Bei gegebenen
Trennbedingungen wird einmal die Probe und einmal der Standard vermessen.
Aus den Chromatogrammen kann die Fläche der Signale bestimmt und
daraus der Gehalt der Probe ermittelt werden.
- Der Korrekturfaktor durch den internen
Standard ergibt sich zu:
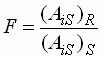
| F |
: |
Korrekturfaktor |
| (AiS)R |
: |
Peakfläche des
internen Standards in der Referenzmessung |
| (AiS)S |
: |
Peakfläche des
internen Standards in der Probemessung |
- Bei der quantitativen Bestimmung sind
zusätzlich Unterschiede im Ansprechverhalten des Detektors auf verschiedene
Substanzen (sog. "response factor") zu berücksichtigen.
Auflösung
- Die Auflösung stellt ein Maß für die Trennung zweier Signale dar. Das DAB berechnet sie nach folgender Formel:
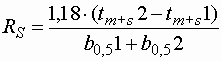
| RS |
: |
Auflösung |
| tm+s |
: |
Gesamtretentionszeit |
| b0,5 |
: |
Peakbreite bei 50 % der Höhe |
- Die Auflösung sollte für quantitative
Bestimmungen größer als 1,4 sein.
Berechnung der Peakfläche
- Die Berechnung der Peakfläche kann nach unterschiedlichen Verfahren vorgenommen werden.
- Nur zur Berechnung symmetrischer Peaks eignet sich die Berechnung der Fläche als Produkt aus der Peakhöhe und der Halbwertsbreite, der Breite des Peaks auf halber Höhe.
- Bei asymmetrischen Peaks kann das Condall-Bosch-Verfahren angewandt werden:
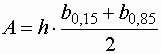
| A |
: |
Peakfläche |
| h |
: |
Höhe des Peaks |
| b0,15 |
: |
Breite bei 15 % der Peakhöhe |
| b0,85 |
: |
Breite bei 85 % der Peakhöhe |
- Ein weiteres Verfahren ist das Ausschneiden und Wiegen der Peaks, das Gewicht ist proportional zur Fläche.
- Heute wird die Berechnung der Peakfläche meist direkt mittels Integration im Messgerät vorgenommen.
Peaksymmetrie
- Im Idealfall folgen die erhaltenen Signale
einer Normalverteilungskurve.
- Dieser Fall ist jedoch sehr selten; meist
zeigen die Signale Abweichungen von der
idealen Symmetrie.
- Steigt der Peak steil an und fällt nach dem
Maximum schwächer ab, so spricht man
von Tailing.
- Ist die ansteigende Signalflanke flacher,
als die abfallende, so nennt man dies Heading oder
Leading.
- Diese Verzerrungen machen eine Auswertung
der Peaks mit Hilfe der einfacheren
Methoden für symmetrische Peaks unmöglich. Hier müssen komplexere
Verfahren angewandt werden.
- Die Symmetrie wird ausgedrückt über
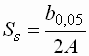
| Ss |
: |
Peaksymmetrie |
| b0,05 |
: |
Peakbreite bei 5 % der
Peakhöhe |
| A |
: |
Breite des Peaks vom
Beginn zu seinem Maximum |
- Ist Ss dabei gleich 1 so ist die Kurve
symmetrisch, Werte über 1 deuten auf Tailing, Werte unter 1 auf Heading hin.
- Im Bereich zwischen 0,8 und 1,2 kann trotz
gegebener Asymmetrie noch ausreichend
genau mit den Auswertmethoden für symmetrische Kurven gearbeitet
werden.
Temperaturprogramm
- Substanzen, die sich unter isothermen
Bedingungen nicht oder nicht hinreichend
genau auftrennen lassen, können evtl. durch Anwendung eines Temperaturprogramms
getrennt werden.
- Hierbei wird die Temperatur der Säule,
gemäß eines festgelegten Programms im Laufe der Analyse erhöht. So
gelangen die leichter flüchtigen Verbindungen bereits zu einem Zeitpunkt in die
Gasphase, zu dem die schwerer flüchtigen aufgrund der zu niedrigen Temperatur noch nicht verdampfen.
- Durch die Wahl eines geeigneten Temperaturprogramms lässt sich die Analysenzeit deutlich verkürzen, bei gleichzeitig besseren Ergebnissen, da die Peaks spitzer und somit besser auswertbar werden.
Anwendungsgebiet
- Blutalkoholbestimmung
- Reinheits- und Gehaltsbestimmungen des DAB
(z.B. Benzin, Vitamin E, etc.)
- Dopinganalyse
- Rückstandsanalytik in Lebensmitteln und
Bedarfsgegenständen
- u.v.m.
Geschichtliches
- Die ersten bekannten Versuche zum Prinzip
der Gaschromatographie gehen auf das 16. Jahrhundert zurück. Im Jahre 1512
versuchte der Straßburger Wundarzt von Brunschwig die destillative Trennung
von Alkohol-Wasser-Mischungen. Er destillierte diese Lösungen durch einen
mit Olivenöl getränkten Schwamm und erhielt reinen Alkohol.
- Erst Mitte des 20. Jahrhunderts wurde die
Gaschromatographie jedoch ein wichtiges analytisches Verfahren, das sowohl
zu präparativer, als auch zu analytischer Trennung von Stoffgemischen
eingesetzt wird.
- Die GC hat mittlerweile Einzug in praktisch
alle analytischen Labors gefunden.
|
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
Demoversion des
pharm@zie-Projektes
vom 27.12.2001. Für den Zugriff auf die aktuelle Online-Version
benötigen Sie ein
persönliches Passwort.
Auch wenn diese Altversion einer Spende eigentlich nicht würdig ist, würde
ich mich natürlich darüber freuen, um die aktuelle Version werbefrei und so
unabhängig wie mir möglich zu halten. Fachliche Unterstützung wäre mir
jedoch noch viel lieber als Geld...
Die Übernahme der Texte und
Bilder in andere On- oder Offline-Angebote ist nur mit schriftlicher
Erlaubnis gestattet!
Datenschutz
|
Impressum
|
© 2000 - 2018
www.BDsoft.de |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|